

Making Better Gene Therapies
Making Better Gene Therapies
Part 1: AAV Foundations

Read this post on the Asimov website. Read Part II.
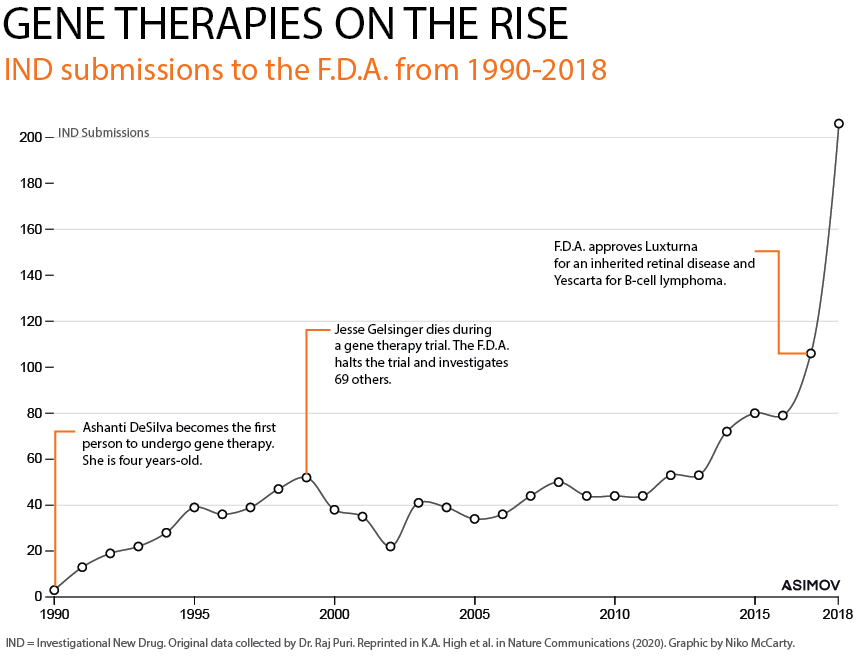
We are living through a gene therapy renaissance. The F.D.A. has approved treatments for spinal muscular atrophy, multiple myeloma, and to restore sight in those with Leber congenital amaurosis, an inherited form of blindness. One-time treatments to lower blood cholesterol are likely on the way.
At least ten thousand people have already received a gene therapy. It is a modern miracle that we can package custom DNA inside of viral vectors, inject them into the body, and treat genetic disorders by supplying cells with “repaired” genes.
Just consider the F.D.A.-approved treatments for hemophilia A and B, genetic conditions that keep blood from clotting. In the early 1900s, most people with the condition died by 13 years of age, partly because there was no way to store blood. The only treatment, at that time, was direct transfusion from a family member. Today, people with hemophilia can be cured after a single injection.
But scattered amongst these success stories are reports of patient deaths and adverse events. An 18-year-old named Jesse Gelsinger was the first to die from a gene therapy, back in 1999. The F.D.A. swiftly shut down the clinical trial and launched investigations into 69 others, kicking off a ‘gene therapy winter.’ Biotech companies shuttered their doors and funding dwindled.
More than twenty years on, the biotech industry has adapted and boomed. Gene therapies are being approved at a rapid clip. Dozens of companies are searching for ways to make them safer and more effective.
In the next blog, we’ll dive into our recent work on using AI-guided design to create more precise gene therapies. But we first need to survey the current landscape: How are gene therapies made, how do they work, and what problems need solving? Only then can we appreciate the problems and perils of delivering genes to cure disease.
Reality
Jesse Gelsinger had a mutation in a single gene, called ornithine transcarbamoylase. This mutation rendered the protein — part of the urea cycle, which turns ammonia into urea — defective, causing ammonia to build up in his bloodstream.
To (hopefully) fix the problem, researchers at the University of Pennsylvania packaged a ‘correct’ version of the OTC gene into an attenuated cold virus — an adenovirus — and injected it into Gelsinger’s artery. He died 4 days later after experiencing “a severe immune reaction to the [viral] vector.”
In the aftermath of Gelsinger’s death, many biotech companies shifted toward adeno-associated viruses, or AAVs, which don’t replicate in human cells on their own, and are not associated with any human disease. An AAV is made from just two parts: a 20-sided icosahedron, called a capsid, and up to 4,700 bases of single-stranded DNA.
AAVs are also naturally diverse. At least 13 serotypes have been discovered so far. Each one has a unique capsid, dotted with sugars and proteins, that together determine which tissues the AAV targets. AAV9 delivers its genetic payload to the liver, muscles, and lungs, whereas AAV2 targets skeletal muscle, retina, liver, and neurons.
If a gene therapy ends up in the wrong places, side effects can ensue. Over 30% of gene therapy trials — including those that use AAVs — report a serious adverse event. Four patients died in a recent trial for X-linked myotubular myopathy (in which muscles slowly waste away) and another died in Pfizer’s trial for Duchenne muscular dystrophy. A 27-year-old named Terry Horgan recently died from a separate gene therapy for Duchenne. An intense immune response against the virus was the likely cause, according to a recent preprint.
An FDA advisory panel recently recommended SRP-9001, an AAV-based gene therapy for Duchenne muscular dystrophy, for approval. This committee doesn’t make final decisions, but the vote was close — 8-to-6 — and the same panel had previously planned to reject the therapy, according to STAT reporting, after a participant’s blood magnesium levels unexpectedly dropped. The F.D.A. paused the trial last June, and it resumed three months later. (Update: The F.D.A. approved the therapy, now called Elevidys, on 22 June 2023.)
To reduce safety issues, AAV gene therapies often target cells in an immune-privileged part of the body (like the eye, which limits inflammatory immune responses “so that vision isn’t harmed by swelling”), or they are given at a low dose.
Luxturna, an AAV gene therapy for blindness, is injected straight into the eye. Other gene therapies that target tissues around the body use a dose as low as 5.8 billion viral genomes per kilogram of body weight (which is not as high as it sounds!) Zolgensma, a one-time therapy for spinal muscular atrophy, is injected into the bloodstream at doses thousands of times higher. Last August, Novartis reported that two patients given Zolgensma died from acute liver failure.
These constraints are problematic. Often, we want to cure more than just genetic forms of blindness, and we often need high doses to do it. Fortunately, a cadre of companies are now engineering AAV capsids, or the DNA inside them, to make safer and more precise gene therapies.
Safety First
To engineer a gene therapy, one first needs to understand how they get made.
Most companies make AAVs by transfecting plasmids, or loops of DNA, into cells. These plasmids encode genes that make functional AAVs. One plasmid encodes the ‘payload’ — usually a human gene, designed to treat disease — which gets coiled up and packaged into the capsid. By tweaking the sequences encoded on these plasmids, one can also alter an AAV’s shell or the payload inside.
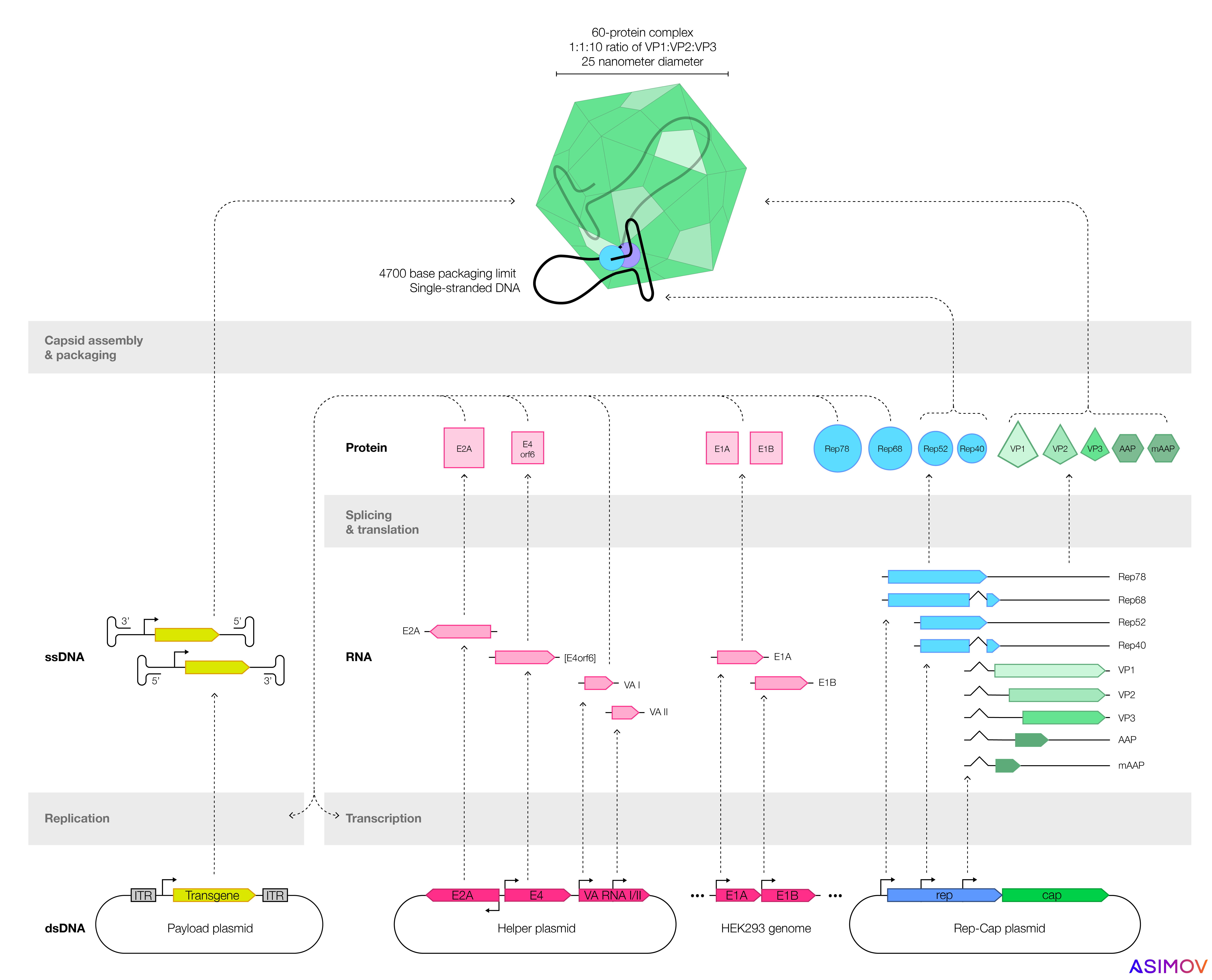
Two types of cells are often used to make AAVs: HEK293 (an immortalized human kidney cell) or SF9, from armyworm moths. But many factors determine a gene therapy's safety and efficacy. We focus here on three: Product quality, immune reactions, and precise expression. All three are also intertwined: lower AAV product quality often means a higher dose, which exacerbates immune reactions.
Let’s look at each problem, one at a time.
The first, product quality, is all about manufacturing. When an immortalized insect or kidney cell starts churning out AAVs, not every capsid gets filled, and not every capsid is created equally. Sometimes the cells make empty protein shells, with no genetic payload. Ideally, the ratio of full-to-empty capsids would be as high as possible, so that a gene therapy’s dose can be kept as low as possible. In nature, AAVs package capsids nearly perfectly, which means almost every particle is infectious. But when a scientist uses recombinant plasmids to produce AAV, the fraction of empties goes up, and nobody seems to fully understand why.
Immortalized insect cells make greater quantities of virus, compared to kidney cells, but the AAVs seem to be less effective at delivering their payloads. Recent work from Nicole Paulk’s group (now the CEO of Siren Biotechnology) suggests two reasons for this: either the insect cells add post-translational modifications — chemical ‘tags’ — to the outer shell of the AAV, or they add epigenetic marks to the single-stranded DNA within. Some of these chemical tags may also cause an immune response. Either way, AAVs made by armyworm moth cells seem to be less potent.
The second problem is immune reactions. Most people are exposed to AAVs during their lives, and more than 60% of adults have antibodies against them. If an AAV capsid is recognized by the body as “foreign,” cytotoxic T lymphocytes fight them off by attacking human cells infected with the virus. Inflammation kicks in and can put a patient at risk. This is why many patients with preexisting antibodies to AAV are excluded from gene therapy trials, according to a recent study. Those antibodies can ‘shut down’ the therapy, and also cause an immune overreaction, which makes a trial less likely to succeed. It also means that lots of people who might need gene therapy can’t get it.
The final problem is precise expression. Remember that AAVs are ‘dumb’ protein shells. They don’t have a brain. They can’t think for themselves. So an AAV floats through the blood and infects many different cells along the way. An AAV designed to target the heart often ends up delivering its DNA to the liver and intestines, too, which can cause side effects later on.
These are the basic problems, then, and each one is connected to all the others. At Asimov, we believe that safer, more precise gene therapies are achievable if we adopt an engineering mindset. We think it’s possible to tweak proteins or alter DNA in juuuuuuust the right way to minimize immune reactions and off-target expression.
And we are not alone. Lots of clever people are coming up with nifty tools to do precisely that.
Engineering Mindset
Each problem — full/empty ratio, immune reactions, and off-target expression — is solvable.
The first problem is partially solvable during the purification process. After cells make AAVs, the capsids can be harvested by centrifugation, for example, which means they’re spun at very high speeds. Heavy things (like filled AAVs) settle to the bottom of the tube, while lighter things (like proteins and water) stay at the top. In this way, it’s also possible to enrich filled capsids, compared to empty ones, by about 50 percent. Another option is to use tangential flow filtration, in which molecules flow past a thin membrane and the small bits get removed. Both methods are imperfect at removing empty capsids.
The immune response and off-target expression problems are more difficult to solve, in part, because they’re so tightly connected. Solving one problem would mitigate the other, and two solutions have risen to the top: Either engineer the AAV capsid to ‘evade’ the immune system or target more precise tissues, or engineer the DNA inside to achieve a similar outcome.
Emeryville-based 4D Molecular Therapeutics engineers AAV capsids. More specifically, they take the plasmids encoding the protein shell and randomly mutate them to make billions of AAV variants. These variants are then screened in monkeys to find capsids that infect cells only in select tissues, like the heart or brain. It seems to be working! Their mutated capsids are being tested in at least five phase I/II clinical trials for everything from eye disorders to Fabry disease, a rare genetic disorder that causes fat build-up in cells.
At the University of California, Berkeley, David Schaffer’s group (a founder of 4D) has helped to unravel how conserved epitopes — a molecule that antibodies bind to — on various AAV serotypes can be mutated to cloak viruses from the immune system. Other groups are also searching for AAV capsids from non-human animals, in nature, that can help gene therapies avoid antibodies in the body.
Other companies, opting for a less “random” approach, are building machine learning tools to design AAV capsids in a more rational way. Dyno Therapeutics recently reported that their AI-designed AAV capsids can pass through the blood-brain barrier in monkeys and deliver genetic payloads 100x better than recombinant AAV9 vectors to the central nervous system.
Besides engineering the AAV shell, one can also tweak DNA directly to help a gene therapy evade the immune system or target precise parts of the body. George Church and Ying Kai Chan at Harvard have crafted AAVs that “evade” the body’s immune and inflammatory responses, simply by adding a short snippet of DNA into the genetic payload. The DNA interacts with toll-like receptor 9, a protein that senses foreign DNA, and dampens its effect.
Other teams are altering DNA to ensure that gene therapies only become active in the ‘correct’ place within the body. A gene therapy for heart disease, in principle, shouldn’t be active in the liver, and a gene therapy for the brain shouldn’t be expressed in the muscle. But this is easier said than done.
A typical recombinant AAV genome has four components: A promoter, the transgene (flanked by short DNA snippets called inverted terminal repeats), and a termination signal. The transgene is the bit of DNA designed to ‘fix’ the broken gene, but the promoter determines how much transgene gets expressed and in which cells.
It is not, contrary to popular belief, always a good idea to crank up transgene levels. Sometimes, the cure for a genetic disease is toxic, which was indeed the case for two gene therapies tested for Rett syndrome in mice and GM2 gangliosidosis, a rare disorder that destroys nerve cells in the brain, in non-human primates. Transgene levels can be tuned by using a ‘weak’ promoter that doesn’t get transcribed into mRNA quite so much, or by packaging AAVs with short, regulatory RNAs that silence DNA.
Figuring out ways to confine transgene expression to specific parts of the body is a more challenging problem. Here, again, there are two common solutions: Either package AAVs with microRNA binding sites that shut down transgene expression in cells with a matching microRNA (a decades-long area of study), or design tissue-specific promoters that only ‘switch on’ in certain cells.
Let’s focus on that second bit: Tissue-specific promoters. There are dozens of them. One, called CD68, confines transgene expression to macrophages, a type of immune cell. Another, called MHCK7, is most active in the heart and muscles. But these naturally-occurring promoters often ‘leak’ into other tissues; when we packaged MHCK7 into AAV9 and injected mice, we found transgene expression not only in heart and muscles, but also in liver, lungs and brain. The problem of making a gene therapy just for the heart or just for the brain, then, remains unsolved.
The process to invent new tissue-specific promoters is also, unfortunately, ridiculously artisanal.
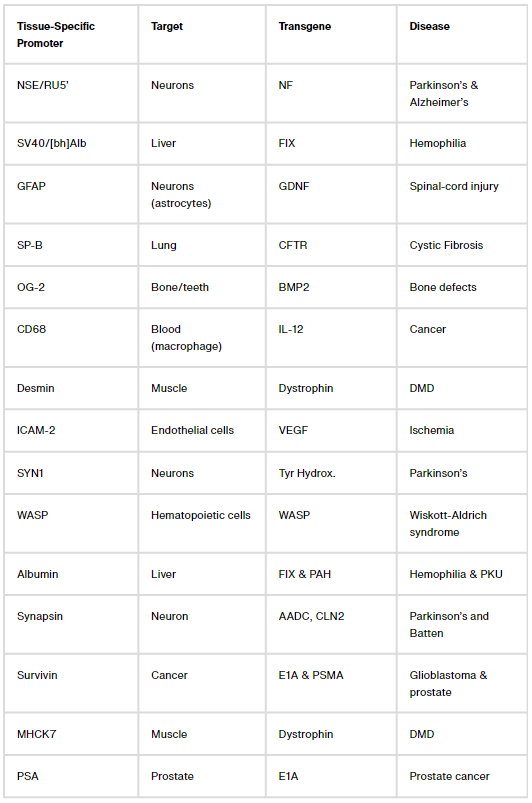
Most tissue-specific promoters are made by smashing a promoter and enhancer (a distant sequence that boosts a promoter’s activity) together. The new sequence is then packed into an AAV and tested in animals. Often, it doesn’t work, because that’s the nature of trial-and-error.
A better way to get tissue-specific expression, we think, is to design promoters that don’t naturally exist in nature. And this has been done before! An MIT group previously made synthetic promoters that target only cancer cells, while avoiding expression in healthy calls. It was impressive work. But cancer cells express lots of genes that healthy cells don’t, and so there are lots of promoters that differentiate the two. It’s challenging to make tissue-specific promoters that distinguish between two healthy cells in the body, like those in the stomach and intestines (which are both made of muscle, after all!)
In the next blog post, we’ll describe how we used an AI transformer model to create 17 synthetic promoters with tissue-specific expression, including one that is 208x more active in the heart than the liver. We think this tool could go a long way toward making gene therapies safer and more precise.
But these are the early days of a new gene therapy era. Jesse Gelsinger was the first person to die from a gene therapy, nearly 25 years ago. And, over the last two decades, biotechnology has made incredible strides. Several gene therapies now have F.D.A. approval and dozens more are likely on the way.
Meanwhile, datasets have ballooned in size. There are millions of genome sequences, transcriptomics datasets, and protein structures. We have atomic-resolution images of AAV capsids. Scientists are tweaking and testing dozens of tissue-specific promoters. All of this progress coincides with a veritable explosion in AI tools. The key will be to bring these advances together to cure once-intractable diseases.
We think that fewer deaths and more success stories will follow, and thus complete this modern miracle.
Join us or subscribe for updates.
Blog Contributors: Scott Estes, Ben Gordon, Mike Leonard, Alec Nielsen, Kevin Smith, Chris Stach, Chris Thorne & Lila Wroblewska. Text by Niko McCarty.
Nice overview!
Very nicely written! I very much enjoyed reading it and looking forward for the next blog post.